zu gewinnen, erhielt ich Präparate mit 5,6 und 5,9 °/o Stickstoff.
Dementsprechend war auch die Ausbeute etwas höher,
als Sch o ll angibt. Das neben dem Chitin vorhandene hemi-
cellulosenähnliche Kohlenhydrat haftet ersterem ziemlich hartnäckig
an und drückt den Stickstoffgehalt der Präparate herunter.
Der Nachweis des Chitins wurde geführt durch Hydrolyse
mit Salzsäure und Darstellung des charakteristischen "salzsauren
Glukosamins.
Hydrolyse von 500 g Pilzrückstand, behufs Gewinnung der Aminosäuren
aus dem Pilzeiweiß.
500 g lufttrockenes Material (mit Äther, Alkohol, und
Wasser extrahiert) mit 7,32 »/o N wurden mit 1500 ccm konzentrierter
Salzsäure übergossen. Die Masse quoll anfangs
auf und wurde im Wasserbade erwärmt, bis fast alles gelöst
war. Die gelbe Flüssigkeit wurde dann vorsichtig am Rückflußkühler
erwärmt, weil bei stärkerem Erhitzen heftiges Schäumen
auftrat. Über Nacht war die Masse zu einer Gallerte erstarrt,
die in 2 Kolben verteilt mit je 750 ccm Salzsäure weitere
8 Stunden zum Sieden erhitzt wurde. Die Hydrolysenflüssigkeit
wurde mit Wasser verdünnt und die reichlich ausgeschiedene
Huminsubstanz abfiltriert. Das dunkelbraune Filtrat wurde im
Vakuum bei 40» eingeengt und blieb 2 Tage über Schwefelsäure
stehen. Dabei schieden sich reichliche Mengen glänzender
Krystallblättchen aus, die nach ümkrystallisieren aus Wasser
die charakteristischen Dodekaederformen des salzsauren Glukosamins
besaßen. Geschmack, Reaktionen und Krystallform
lassen keinen Zweifel an der Natur dieses Körpers zu, der
jedenfalls aus dem Pilzchitin entstanden war. Insgesamt wurden
20 g erhalten.
Die dickflüssige Mutterlauge wurde weiter eingeengt, bis
sich zähe Blasen bildeten, mit 2 Volumina absolutem Alkohol
übergossen und mit trockenem Salzsäuregas gesättigt. Die
Lösung wurde bei vermindertem Druck eingeengt und die Veresterung
wiederholt. Die Isolierung der Aminosäureester ge-
schah nach E. F is c h e r ') mit Hilfe von Natriumhydroxyd und
•) Diese Zeitschrift, Bd. 3H, S. 151.
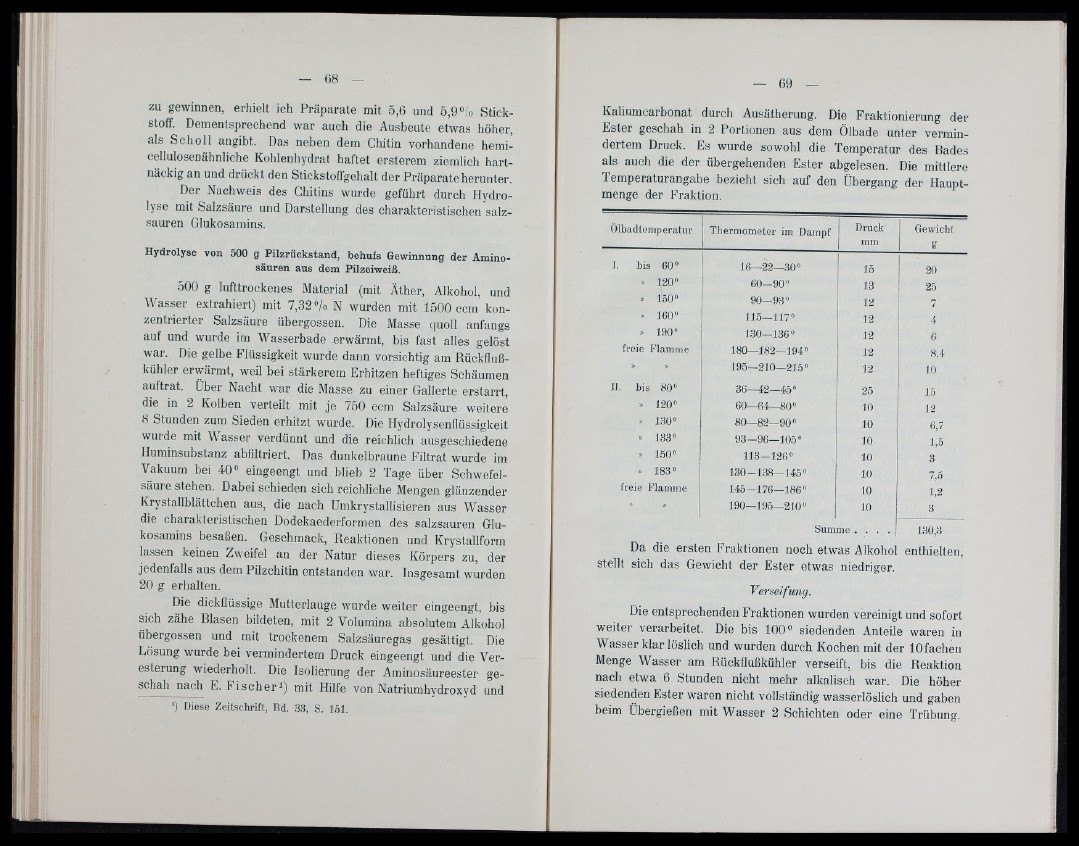
Kaliumcarbonat durch Ausätherung. Die Fraktionierung der
Ester geschah in 2 Portionen aus dem Ölbade unter vermindertem
Druck. Es wurde sowohl die Temperatur des Bades
als auch die der übergehenden Ester abgelesen. Die mittlere
Temperaturangabe bezieht sich auf den Übergang der Hauptmenge
der Fraktion.
Olbadtemperatur Thermometer im Dampf Druck
mm
Gewicht
g
I. bis 60» 16—22—.30» 15 20
» 120» 60—90» 13 25
» 150» 90—93« 12 7
» 160» 115—117« 12 4
. 190» 130—136» 12 6
freie Flamme 1 8 0 -1 8 2 -1 9 4 » 12 8.4
» » 195—210—215» 12 10
II. bis 80» 3 6 -4 2—45» 25 15
» 120» 60 64 80» 10 12
» 1,30» 80—82—90» 10 6,7
» 133» 93—96—105» 10 1,5
» 150» 113-126» 10 3
» 183» 1 30-138—145» 10 7,5
freie Flamme 1 4 5 -1 7 6—186» 10 1,2
190—195—210» 10 3
Summe . . . . 1.30,3
Da die ersten Fraktionen noch etwas Alkohol enthielten,
stellt sich das Gewicht der Ester etwas niedriger.
Verseifung.
Die entsprechenden Fraktionen wurden vereinigt und sofort
weiter verarbeitet. Die bis 100» siedenden Anteile waren in
Wasser klar löslich und wurden durch Kochen mit der lOfachen
Menge Wasser am Rückflußkühler verseift, bis die Reaktion
nach etwa 6 Stunden nicht mehr alkalisch war. Die höher
siedenden Ester waren nicht vollständig wasserlöslich und gaben
beim Übergießen mit Wasser 2 Schichten oder eine Trübung.